Genômica
Genomics
Genômica é um campo interdisciplinar da biologia com foco na estrutura, função, evolução, mapeamento e edição de genomas. Um genoma é o conjunto completo de DNA de um organismo, incluindo todos os seus genes. Em contraste com a genética, que se refere ao estudo de genes individuais e seus papéis na herança, a genômica visa a caracterização e quantificação coletiva de todos os genes de um organismo, suas inter-relações e influência no organismo. Os genes podem direcionar a produção de proteínas com o auxílio de enzimas e moléculas mensageiras. Por sua vez, as proteínas constituem as estruturas do corpo, como órgãos e tecidos, além de controlar as reações químicas e transportar sinais entre as células. A genômica também envolve o sequenciamento e a análise de genomas por meio do uso de sequenciamento de DNA de alto rendimento e bioinformática para montar e analisar a função e a estrutura de genomas inteiros. Os avanços na genômica desencadearam uma revolução na pesquisa baseada em descobertas e na biologia de sistemas para facilitar a compreensão até mesmo dos sistemas biológicos mais complexos, como o cérebro.
O campo também inclui estudos de fenômenos intragenômicos (dentro do genoma) como epistasia (efeito de um gene em outro), pleiotropia (um gene afetando mais de uma característica), heterose (vigor híbrido) e outras interações entre loci e alelos dentro do genoma.
Conteúdo
História
Etimologia
Do grego ΓΕΝ gen , "gene" (gama, épsilon, nu, épsilon) que significa "tornar-se, criar, criação, nascimento "e variantes subsequentes: genealogia, gênese, genética, genética, genômero, genótipo, gênero etc. Enquanto a palavra genoma (do alemão Genom , atribuído a Hans Winkler) estava em uso em inglês já em 1926, o termo genômica foi cunhado por Tom Roderick, um geneticista do Jackson Laboratory (Bar Harbor, Maine), durante uma reunião realizada Eu em Maryland no mapeamento do genoma humano em 1986.
Esforços iniciais de sequenciamento
Após a confirmação de Rosalind Franklin da estrutura helicoidal do DNA, a publicação de James D. Watson e Francis Crick do estrutura do DNA em 1953 e a publicação de Fred Sanger da sequência de aminoácidos da insulina em 1955, o sequenciamento do ácido nucléico tornou-se um dos principais alvos dos primeiros biólogos moleculares. Em 1964, Robert W. Holley e colegas publicaram a primeira sequência de ácido nucleico já determinada, a sequência de ribonucleotídeos do RNA de transferência de alanina. Estendendo este trabalho, Marshall Nirenberg e Philip Leder revelaram a natureza tripla do código genético e foram capazes de determinar as sequências de 54 dos 64 códons em seus experimentos. Em 1972, Walter Fiers e sua equipe do Laboratório de Biologia Molecular da Universidade de Ghent (Ghent, Bélgica) foram os primeiros a determinar a sequência de um gene: o gene da proteína capsidial do bacteriófago MS2. O grupo de Fiers expandiu seu trabalho de proteína de revestimento MS2, determinando a sequência de nucleotídeos completa do bacteriófago MS2-RNA (cujo genoma codifica apenas quatro genes em 3569 pares de bases) e do vírus Simian 40 em 1976 e 1978, respectivamente.
Tecnologia de sequenciamento de DNA desenvolvida
Além de seu trabalho seminal sobre a sequência de aminoácidos da insulina, Frederick Sanger e seus colegas desempenharam um papel fundamental no desenvolvimento de técnicas de sequenciamento de DNA que permitiram o estabelecimento de projetos de sequenciamento de genoma. Em 1975, ele e Alan Coulson publicaram um procedimento de sequenciamento usando DNA polimerase com nucleotídeos radiomarcados que ele chamou de Mais e menos técnica . Isso envolveu dois métodos intimamente relacionados que geraram oligonucleotídeos curtos com terminais 3 'definidos. Estes podem ser fracionados por eletroforese em um gel de poliacrilamida (chamada eletroforese em gel de poliacrilamida) e visualizados por autoradiografia. O procedimento podia sequenciar até 80 nucleotídeos de uma vez e era uma grande melhoria, mas ainda era muito trabalhoso. Não obstante, em 1977 seu grupo foi capaz de sequenciar a maioria dos 5.386 nucleotídeos do bacteriófago de fita simples φX174, completando o primeiro genoma baseado em DNA totalmente sequenciado. O refinamento do método Mais e Menos resultou na terminação da cadeia, ou método Sanger (veja abaixo), que formou a base das técnicas de sequenciamento de DNA, mapeamento de genoma, armazenamento de dados e análise bioinformática mais amplamente utilizado no quarto século seguinte de pesquisa. No mesmo ano, Walter Gilbert e Allan Maxam da Universidade de Harvard desenvolveram independentemente o método Maxam-Gilbert (também conhecido como o método químico ) de sequenciamento de DNA, envolvendo a clivagem preferencial de DNA em bases conhecidas, método eficiente. Por seu trabalho inovador no sequenciamento de ácidos nucléicos, Gilbert e Sanger dividiram metade do Prêmio Nobel de Química em 1980 com Paul Berg (DNA recombinante).
Genomas completos
O advento dessas tecnologias resultou em uma rápida intensificação do escopo e da velocidade de conclusão dos projetos de sequenciamento do genoma. A primeira sequência completa do genoma de uma organela eucariótica, a mitocôndria humana (16.568 bp, cerca de 16,6 kb), foi relatada em 1981, e os primeiros genomas do cloroplasto se seguiram em 1986. Em 1992, o primeiro cromossomo eucariótico, cromossomo III da levedura de cerveja Saccharomyces cerevisiae (315 kb) foi sequenciado. O primeiro organismo de vida livre a ser sequenciado foi o de Haemophilus influenzae (1,8 Mb) em 1995. No ano seguinte, um consórcio de pesquisadores de laboratórios da América do Norte, Europa e Japão anunciou a conclusão do primeira sequência completa do genoma de um eucarioto, S. cerevisiae (12,1 Mb), e desde então os genomas continuaram sendo sequenciados em um ritmo de crescimento exponencial. Em outubro de 2011, as sequências completas estavam disponíveis para: 2.719 vírus, 1.115 arquéias e bactérias e 36 eucariotos, dos quais cerca de metade são fungos.
A maioria dos microrganismos cujos genomas foram completamente sequenciados são problemáticos patógenos, como Haemophilus influenzae , que resultou em um viés pronunciado em sua distribuição filogenética em comparação com a amplitude da diversidade microbiana. Das outras espécies sequenciadas, a maioria foi escolhida porque eram organismos modelo bem estudados ou prometiam se tornar bons modelos. A levedura ( Saccharomyces cerevisiae ) tem sido um organismo modelo importante para a célula eucariótica, enquanto a mosca da fruta Drosophila melanogaster tem sido uma ferramenta muito importante (especialmente no início do período pré-molecular genética). O verme Caenorhabditis elegans é um modelo simples frequentemente usado para organismos multicelulares. O peixe-zebra Brachydanio rerio é usado para muitos estudos de desenvolvimento no nível molecular, e a planta Arabidopsis thaliana é um organismo modelo para plantas com flores. O baiacu japonês ( Takifugu rubripes ) e o baiacu verde manchado ( Tetraodon nigroviridis ) são interessantes por causa de seus genomas pequenos e compactos, que contêm muito pouco DNA não codificador em comparação com a maioria das espécies . O cão mamífero ( Canis familiaris ), rato marrom ( Rattus norvegicus ), camundongo ( Mus musculus ) e chimpanzé ( Pan troglodytes ) são animais modelo importantes na pesquisa médica.
Um rascunho do genoma humano foi concluído pelo Projeto Genoma Humano no início de 2001, criando muito alarde. Este projeto, concluído em 2003, sequenciou todo o genoma de uma pessoa específica e, em 2007, essa sequência foi declarada "concluída" (menos de um erro em 20.000 bases e todos os cromossomos reunidos). Nos anos desde então, os genomas de muitos outros indivíduos foram sequenciados, em parte sob os auspícios do Projeto 1000 Genomes, que anunciou o sequenciamento de 1.092 genomas em outubro de 2012. A conclusão deste projeto foi possibilitada pelo desenvolvimento de mais tecnologias de sequenciamento eficientes e exigiram o compromisso de recursos de bioinformática significativos de uma grande colaboração internacional. A análise contínua de dados genômicos humanos tem profundas repercussões políticas e sociais para as sociedades humanas.
A revolução "ômicas"
O neologismo da língua inglesa ômicas refere-se informalmente a um campo de estudo em biologia terminando em -omics , como genômica, proteômica ou metabolômica. O sufixo relacionado -ome é usado para endereçar os objetos de estudo de tais campos, como o genoma, proteoma ou metaboloma, respectivamente. O sufixo -ome , conforme usado em biologia molecular, se refere a uma totalidade de algum tipo; da mesma forma, ômica passou a se referir geralmente ao estudo de grandes e abrangentes conjuntos de dados biológicos. Embora o crescimento no uso do termo tenha levado alguns cientistas (Jonathan Eisen, entre outros) a alegar que ele foi sobrevendido, ele reflete a mudança na orientação em direção à análise quantitativa de sortimento completo ou quase completo de todos os constituintes de um sistema. No estudo das simbioses, por exemplo, os pesquisadores que antes se limitavam ao estudo de um único produto gênico podem agora comparar simultaneamente o complemento total de vários tipos de moléculas biológicas.
Análise do genoma
Depois que um organismo foi selecionado, os projetos de genoma envolvem três componentes: o sequenciamento do DNA, a montagem dessa sequência para criar uma representação do cromossomo original e a anotação e análise dessa representação.
Sequenciamento
Historicamente, o sequenciamento era feito em centros de sequenciamento , instalações centralizadas (variando de grandes instituições independentes, como o Joint Genome Institute, que sequenciam dezenas de terabases por ano, até instalações centrais locais de biologia molecular) que contêm pesquisas laboratórios com instrumentação dispendiosa e suporte técnico necessário. Conforme a tecnologia de sequenciamento continua a melhorar, no entanto, uma nova geração de sequenciadores de bancada de retorno rápido e eficazes está ao alcance do laboratório acadêmico médio. No geral, as abordagens de sequenciamento do genoma se enquadram em duas categorias amplas, shotgun e sequenciamento de alta capacidade (ou próxima geração ).
O sequenciamento Shotgun é um método de sequenciamento desenvolvido para a análise de sequências de DNA com mais de 1000 pares de bases, até e incluindo cromossomos inteiros. É nomeado por analogia com o padrão de disparo quase aleatório de rápida expansão de uma espingarda. Uma vez que o sequenciamento de eletroforese em gel só pode ser usado para sequências razoavelmente curtas (100 a 1000 pares de bases), sequências de DNA mais longas devem ser quebradas em pequenos segmentos aleatórios que são então sequenciados para obter leituras . Múltiplas leituras sobrepostas para o DNA alvo são obtidas realizando várias rodadas desta fragmentação e sequenciamento. Os programas de computador então usam as extremidades sobrepostas de diferentes leituras para montá-las em uma sequência contínua. O sequenciamento Shotgun é um processo de amostragem aleatório, exigindo amostragem excessiva para garantir que um determinado nucleotídeo seja representado na sequência reconstruída; o número médio de leituras pelas quais um genoma é super amostrado é conhecido como cobertura.
Durante grande parte de sua história, a tecnologia subjacente ao sequenciamento shotgun foi o método clássico de terminação em cadeia ou 'método Sanger', que se baseia na incorporação seletiva de didesoxinucleotídeos de terminação de cadeia pela DNA polimerase durante a replicação de DNA in vitro. Recentemente, o sequenciamento shotgun foi suplantado por métodos de sequenciamento de alto rendimento, especialmente para análises de genoma automatizadas em grande escala. No entanto, o método Sanger continua em amplo uso, principalmente para projetos de menor escala e para obter leituras de sequências de DNA contíguas especialmente longas (& gt; 500 nucleotídeos). Os métodos de terminação de cadeia requerem um molde de DNA de fita simples, um primer de DNA, uma DNA polimerase, desoxinucleosidotrifosfatos normais (dNTPs) e nucleotídeos modificados (didesoxiNTPs) que terminam o alongamento do DNA. Estes nucleotídeos de terminação de cadeia carecem de um grupo 3'-OH necessário para a formação de uma ligação fosfodiéster entre dois nucleotídeos, fazendo com que a DNA polimerase cesse a extensão do DNA quando um ddNTP é incorporado. Os ddNTPs podem ser marcados radioativamente ou fluorescentemente para detecção em sequenciadores de DNA. Normalmente, essas máquinas podem sequenciar até 96 amostras de DNA em um único lote (execução) em até 48 execuções por dia.
A alta demanda por sequenciamento de baixo custo impulsionou o desenvolvimento de sequenciamento de alto rendimento tecnologias que paralelizam o processo de sequenciamento, produzindo milhares ou milhões de sequências de uma vez. O sequenciamento de alto rendimento tem como objetivo reduzir o custo do sequenciamento de DNA além do que é possível com os métodos padrão de terminação de corante. No sequenciamento de ultra-alto rendimento, até 500.000 operações de sequenciamento por síntese podem ser executadas em paralelo.
O método de sequenciamento de corante Illumina é baseado em terminadores de corante reversíveis e foi desenvolvido em 1996 no Instituto de Pesquisa Biomédica de Genebra, de Pascal Mayer e Laurent Farinelli. Neste método, moléculas de DNA e primers são primeiro anexados em uma lâmina e amplificados com polimerase para que colônias clonais locais, inicialmente denominadas "colônias de DNA", sejam formadas. Para determinar a sequência, quatro tipos de bases terminadoras reversíveis (bases RT) são adicionadas e os nucleotídeos não incorporados são lavados. Ao contrário do pirosequenciamento, as cadeias de DNA são estendidas um nucleotídeo por vez e a aquisição de imagem pode ser realizada em um momento atrasado, permitindo que arranjos muito grandes de colônias de DNA sejam capturados por imagens sequenciais tiradas de uma única câmera. A dissociação da reação enzimática e da captura de imagem permite um rendimento ideal e capacidade de sequenciamento teoricamente ilimitada; com uma configuração ideal, o rendimento final do instrumento depende apenas da taxa de conversão A / D da câmera. A câmera tira imagens dos nucleotídeos marcados com fluorescência, então o corante junto com o bloqueador terminal 3 'é quimicamente removido do DNA, permitindo o próximo ciclo.
Uma abordagem alternativa, o sequenciamento de semicondutores de íons, é baseada na química de replicação de DNA padrão. Essa tecnologia mede a liberação de um íon de hidrogênio cada vez que uma base é incorporada. Um micropoço contendo DNA molde é inundado com um único nucleotídeo; se o nucleotídeo for complementar à fita molde, ele será incorporado e um íon de hidrogênio será liberado. Esta versão aciona um sensor de íons ISFET. Se um homopolímero estiver presente na sequência modelo, vários nucleotídeos serão incorporados em um único ciclo de inundação e o sinal elétrico detectado será proporcionalmente maior.
Montagem
A montagem da sequência refere-se ao alinhamento e fusão de fragmentos de uma sequência de DNA muito mais longa para reconstruir a sequência original. Isso é necessário porque a tecnologia de sequenciamento de DNA atual não consegue ler genomas inteiros como uma sequência contínua, mas sim pequenos pedaços entre 20 e 1000 bases, dependendo da tecnologia usada. As tecnologias de sequenciação de terceira geração, como PacBio ou Oxford Nanopore, geram rotineiramente leituras de sequenciamento & gt; 10 kb de comprimento; no entanto, eles têm uma alta taxa de erro de aproximadamente 15%. Normalmente, os fragmentos curtos, chamados de leituras, resultam do sequenciamento shotgun de DNA genômico ou transcrições de genes (ESTs).
A montagem pode ser categorizada em duas abordagens: montagem de novo , para genomas que não são semelhantes a nenhum sequenciado no passado e montagem comparativa, que usa a sequência existente de um organismo intimamente relacionado como referência durante a montagem. Em relação à montagem comparativa, a montagem de novo é computacionalmente difícil (NP-difícil), tornando-a menos favorável para tecnologias NGS de leitura curta. Dentro do paradigma de montagem de novo , há duas estratégias principais para montagem, estratégias de caminho Euleriano e estratégias de consenso de layout de sobreposição (OLC). As estratégias OLC, em última análise, tentam criar um caminho hamiltoniano através de um gráfico de sobreposição que é um problema NP-difícil. As estratégias de caminho Euleriano são computacionalmente mais tratáveis porque tentam encontrar um caminho Euleriano através de um gráfico deBruijn.
Os genomas acabados são definidos como tendo uma única sequência contígua sem ambigüidades representando cada réplica.
Anotação
A montagem da sequência de DNA sozinha tem pouco valor sem análise adicional. A anotação do genoma é o processo de anexar informações biológicas a sequências e consiste em três etapas principais:
- identificar porções do genoma que não codificam para proteínas
- identificar elementos em o genoma, um processo chamado predição de gene, e
- anexando informações biológicas a esses elementos.
Ferramentas de anotação automática tentam realizar essas etapas in silico , em oposição à anotação manual (também conhecida como curadoria), que envolve conhecimento humano e verificação experimental potencial. Idealmente, essas abordagens coexistem e se complementam no mesmo pipeline de anotação (também veja abaixo).
Tradicionalmente, o nível básico de anotação é usar o BLAST para encontrar semelhanças e, em seguida, anotar genomas com base em homólogos . Mais recentemente, informações adicionais foram adicionadas à plataforma de anotação. As informações adicionais permitem que anotadores manuais deconvoluem discrepâncias entre genes que recebem a mesma anotação. Alguns bancos de dados usam informações de contexto do genoma, pontuações de similaridade, dados experimentais e integrações de outros recursos para fornecer anotações do genoma por meio de sua abordagem de subsistemas. Outros bancos de dados (por exemplo, Ensembl) contam com fontes de dados com curadoria, bem como uma gama de ferramentas de software em seu pipeline de anotação de genoma automatizado. A anotação estrutural consiste na identificação de elementos genômicos, principalmente ORFs e sua localização, ou estrutura do gene. A anotação funcional consiste em anexar informações biológicas aos elementos genômicos.
Sequenciamento de pipelines e bancos de dados
A necessidade de reprodutibilidade e gerenciamento eficiente da grande quantidade de dados associados com projetos de genoma significa que pipelines computacionais têm aplicações importantes em genômica.
Áreas de pesquisa
Genômica funcional
A genômica funcional é um campo da biologia molecular que tenta fazer uso da vasta riqueza de dados produzidos por projetos genômicos (como projetos de sequenciamento de genoma) para descrever funções e interações de genes (e proteínas). A genômica funcional concentra-se nos aspectos dinâmicos, como a transcrição do gene, a tradução e as interações proteína-proteína, em oposição aos aspectos estáticos da informação genômica, como sequência ou estruturas de DNA. A genômica funcional tenta responder a perguntas sobre a função do DNA nos níveis dos genes, transcrições de RNA e produtos proteicos. Uma característica fundamental dos estudos de genômica funcional é a abordagem de todo o genoma para essas questões, geralmente envolvendo métodos de alto rendimento, em vez de uma abordagem mais tradicional "gene por gene".
Um ramo importante da genômica é ainda preocupada com o sequenciamento de genomas de vários organismos, mas o conhecimento de genomas completos criou a possibilidade para o campo da genômica funcional, principalmente preocupada com padrões de expressão gênica durante várias condições. As ferramentas mais importantes aqui são microarrays e bioinformática.
Genômica estrutural
A genômica estrutural busca descrever a estrutura tridimensional de cada proteína codificada por um determinado genoma. Esta abordagem baseada em genoma permite um método de alto rendimento de determinação de estrutura por uma combinação de abordagens experimentais e de modelagem. A principal diferença entre a genômica estrutural e a previsão estrutural tradicional é que a genômica estrutural tenta determinar a estrutura de cada proteína codificada pelo genoma, em vez de se concentrar em uma proteína específica. Com as sequências do genoma completo disponíveis, a previsão da estrutura pode ser feita mais rapidamente por meio de uma combinação de abordagens experimentais e de modelagem, especialmente porque a disponibilidade de um grande número de genomas sequenciados e estruturas de proteínas previamente resolvidas permite que os cientistas modelem a estrutura da proteína nas estruturas de homólogos. A genômica estrutural envolve a adoção de um grande número de abordagens para a determinação da estrutura, incluindo métodos experimentais usando sequências genômicas ou abordagens baseadas em modelagem com base na sequência ou homologia estrutural com uma proteína de estrutura conhecida ou com base em princípios químicos e físicos para uma proteína sem homologia com qualquer estrutura conhecida. Ao contrário da biologia estrutural tradicional, a determinação da estrutura de uma proteína por meio de um esforço de genômica estrutural frequentemente (mas nem sempre) vem antes de se saber qualquer coisa sobre a função da proteína. Isso levanta novos desafios na bioinformática estrutural, ou seja, determinar a função da proteína a partir de sua estrutura 3D.
Epigenômica
A epigenômica é o estudo do conjunto completo de modificações epigenéticas no material genético de uma célula , conhecido como epigenoma. Modificações epigenéticas são modificações reversíveis no DNA ou histonas de uma célula que afetam a expressão do gene sem alterar a sequência do DNA (Russell 2010 p. 475). Duas das modificações epigenéticas mais caracterizadas são a metilação do DNA e a modificação das histonas. As modificações epigenéticas desempenham um papel importante na expressão e regulação gênica e estão envolvidas em vários processos celulares, como na diferenciação / desenvolvimento e tumorigênese. O estudo da epigenética em nível global só foi possível recentemente através da adaptação de ensaios genômicos de alto rendimento.
Metagenômica
Metagenômica é o estudo de metagenomas , material genético recuperado diretamente de amostras ambientais. O amplo campo também pode ser referido como genômica ambiental, ecogenômica ou genômica comunitária. Enquanto a microbiologia tradicional e o sequenciamento do genoma microbiano dependem de culturas clonais cultivadas, o sequenciamento de genes ambientais iniciais clonou genes específicos (geralmente o gene 16S rRNA) para produzir um perfil de diversidade em uma amostra natural. Esse trabalho revelou que a grande maioria da biodiversidade microbiana foi perdida por métodos baseados em cultivo. Estudos recentes usam sequenciamento Sanger "shotgun" ou pirosequenciamento maciçamente paralelo para obter amostras amplamente imparciais de todos os genes de todos os membros das comunidades amostradas. Devido ao seu poder de revelar a diversidade anteriormente oculta da vida microscópica, a metagenômica oferece uma lente poderosa para visualizar o mundo microbiano que tem o potencial de revolucionar a compreensão de todo o mundo vivo.
Sistemas modelo
Os bacteriófagos desempenharam e continuam a desempenhar um papel fundamental na genética bacteriana e na biologia molecular. Historicamente, eles foram usados para definir a estrutura e a regulação do gene. Além disso, o primeiro genoma a ser sequenciado foi um bacteriófago. No entanto, a pesquisa de bacteriófagos não liderou a revolução genômica, que é claramente dominada pela genômica bacteriana. Só muito recentemente o estudo dos genomas de bacteriófagos se tornou proeminente, permitindo assim aos pesquisadores compreender os mecanismos subjacentes à evolução do fago. As sequências do genoma do bacteriófago podem ser obtidas por meio do sequenciamento direto de bacteriófagos isolados, mas também podem ser derivadas como parte de genomas microbianos. A análise de genomas bacterianos mostrou que uma quantidade substancial de DNA microbiano consiste em sequências de profagos e elementos semelhantes a profagos. Uma mineração detalhada do banco de dados dessas sequências oferece insights sobre o papel dos profagos na formação do genoma bacteriano: No geral, este método verificou muitos grupos bacteriófagos conhecidos, tornando-o uma ferramenta útil para prever as relações dos profagos nos genomas bacterianos.
No momento, existem 24 cianobactérias para as quais uma sequência total do genoma está disponível. 15 dessas cianobactérias vêm do meio marinho. Estas são seis cepas Prochlorococcus , sete cepas marinhas Synechococcus , Trichodesmium erythraeum IMS101 e Crocosphaera watsonii WH8501. Vários estudos têm demonstrado como essas sequências podem ser usadas com muito sucesso para inferir características ecológicas e fisiológicas importantes de cianobactérias marinhas. No entanto, existem muitos outros projetos de genoma em andamento, entre os quais existem outros Prochlorococcus e isolados marinhos de Synechococcus , Acaryochloris e Prochloron , as cianobactérias filamentosas fixadoras de N2 Nodularia spumigena , Lyngbya aestuarii e Lyngbya majuscula , bem como bacteriófagos que infectam cianobacerias marinhas. Assim, o crescente corpo de informações do genoma também pode ser aproveitado de uma maneira mais geral para tratar de problemas globais por meio da aplicação de uma abordagem comparativa. Alguns exemplos novos e estimulantes de progresso neste campo são a identificação de genes para RNAs reguladores, insights sobre a origem evolutiva da fotossíntese ou estimativa da contribuição da transferência horizontal de genes para os genomas que foram analisados.
Aplicações da genômica
A genômica forneceu aplicações em muitos campos, incluindo medicina, biotecnologia, antropologia e outras ciências sociais.
Medicina genômica
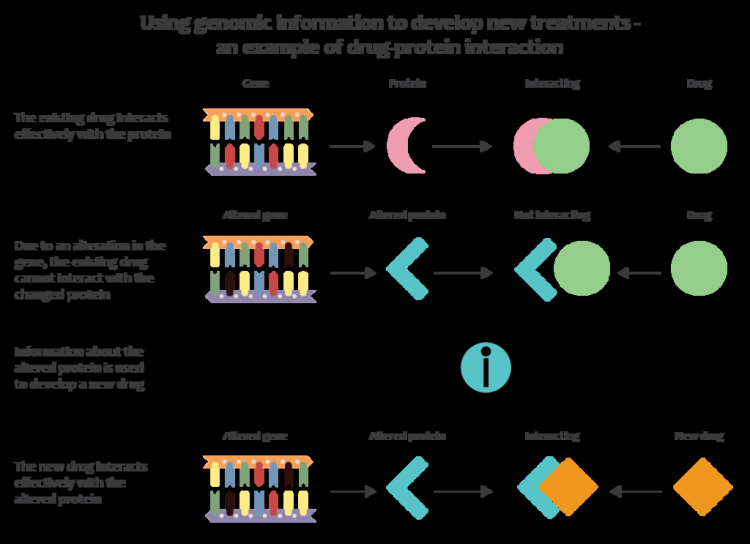
Genômica de última geração as tecnologias permitem que os médicos e pesquisadores biomédicos aumentem drasticamente a quantidade de dados genômicos coletados em grandes populações de estudo. Quando combinado com novas abordagens de informática que integram muitos tipos de dados com dados genômicos na pesquisa de doenças, isso permite que os pesquisadores entendam melhor as bases genéticas da resposta a medicamentos e doenças. Os primeiros esforços para aplicar o genoma à medicina incluíram os de uma equipe de Stanford liderada por Euan Ashley, que desenvolveu as primeiras ferramentas para a interpretação médica de um genoma humano. Por exemplo, o programa de pesquisa Todos nós visa coletar dados da sequência do genoma de 1 milhão de participantes para se tornar um componente crítico da plataforma de pesquisa da medicina de precisão.
Biologia sintética e bioengenharia
O crescimento do conhecimento genômico permitiu aplicações cada vez mais sofisticadas da biologia sintética. Em 2010, pesquisadores do J. Craig Venter Institute anunciaram a criação de uma espécie parcialmente sintética de bactéria, Mycoplasma laboratorium , derivada do genoma de Mycoplasma genitalium .
Genômica da conservação
Os conservacionistas podem usar as informações coletadas pelo sequenciamento genômico para avaliar melhor os fatores genéticos essenciais para a conservação das espécies, como a diversidade genética de uma população ou se um indivíduo é heterozigoto para um recessivo desordem genética hereditária. Ao usar dados genômicos para avaliar os efeitos dos processos evolutivos e detectar padrões de variação em uma determinada população, os conservacionistas podem formular planos para ajudar uma determinada espécie sem tantas variáveis desconhecidas quanto aquelas não abordadas por abordagens genéticas padrão.
Genética
Genética A genética é um ramo da biologia que se preocupa com o estudo dos …

George W. Citroner
George W. Citroner cobre as últimas notícias em medicina e saúde. Ele aparece em …
